
Hosted by Sophie Ward & Tom Reeves

Transcription
Imagine sitting in this um this sterile, fluorescent lit conference room in Washington D.C.
Right, a very high stakes room.
Exactly. You are looking at a panel of the world's most elite neurologists, statisticians, researchers. These are the independent experts hand-picked by the FDA.
And they're there to review a highly anticipated new drug.
Yeah, a proposed treatment for a disease that has just terrorized humanity for decades. And one by one, they cast their votes on whether this drug should be approved.
The tension must have been incredible.
Oh, absolutely. And the vote comes back 10 to 0 against approval.
Unanimous.
Yeah.
With like one person voting uncertain, a resounding no.
Which is pretty rare for it to be that definitive.
It is. So, what does the regulatory agency do? They approve it anyway. They just push it right through against the experts.
It's still hard to believe that actually happened.
I know, right? That genuinely unprecedented moment is just one chapter in what is arguably the most dramatic medical detective story of the last century. So, welcome to today's deep dive.
It really is the perfect starting point. I mean, that specific controversy encapsulates everything about this field.
The desperation, right?
Exactly. The desperation, the biological complexity, and this incredible tension between scientific rigor and well, human hope.
Yeah, human hope is a powerful thing.
It is. We are examining an area of medicine that has absorbed billions of dollars in research funding and defined countless brilliant careers, only to yield a massive graveyard of failed clinical trials.
Which brings us to the core of our analysis today. We are dissecting the historical and biological evolution of the amyloid hypothesis.
The Holy Grail of Alzheimer's research.
The absolute Holy Grail. We want to understand if this 30-year-old biological theory has finally been validated.
And crucially, what that validation actually means in the real world.
Right. What does it mean for patients, for doctors, and for global health care economics? So, to you listening, whether you're deep in the weeds of health care infrastructure or tracking pharma pipelines or maybe you're just trying to separate the genuine scientific breakthroughs from, you know, those sensationalized media headlines.
There are a lot of those headlines out there.
So many. We are going to cut through that and break down the exact cellular mechanisms and clinical data driving this paradigm shift.
It's essential that we approach this with a very specific lens, though.
How so?
Well, the transition from a statistically significant outcome in a highly controlled phase three trial to the messy logistical reality of global health care is incredibly fraught.
It's not just a plug-and-play situation.
Not at all. We have reached a definitive milestone in neurology, yes, but applying that milestone to a population scale introduces systemic hurdles that are going to test medical infrastructures to their absolute limits.
Okay, to understand the magnitude of where we are right now, we really have to trace the biology back to its origin.
Into the dark ages of Alzheimer's research.
Exactly. Let's wind the clock back to 1992. The Journal Science publishes a landmark paper by John Hardy and Gerald Higgins.
A massively influential paper.
Right, they introduced the amyloid cascade hypothesis. And the foundational logic they laid out was incredibly elegant.
It made perfect sense at the time.
You look at the brain of a patient with Alzheimer's and you see these dense, sticky plaques built up between the neurons.
And those plaques are made of a protein fragment called amyloid beta.
Yeah. So, the hypothesis was straightforward, like amyloid buildup is the primary pathogenic trigger. It's the first domino to fall.
Clear the amyloid, stop the disease progression. That was the mantra.
Right.
And the elegance of that hypothesis is exactly why it captured the entire field of neuroscience for three decades. And I should add, it wasn't just a blind guess based on pathology.
It had a lot of backing.
It was deeply rooted in genetics.
Hardy and Higgins looked at patients with early onset familial Alzheimer's.
People developing the disease in their 40s and 50s, which is just tragic.
Devastating.
And they found that these patients had specific mutations in the gene that produces the amyloid precursor protein or APP.
So, the smoking gun was basically built into their DNA.
Precisely. They also looked at Down syndrome patients who carry an extra copy of chromosome 21.
Which happens to be exactly where the APP gene is located, right?
Exactly. And these individuals almost universally develop Alzheimer's pathology as they age. So, the genetic evidence practically screamed that amyloid was the primary culprit.
Okay, but I want to push back on that a bit because tracing the line from we found the genetic trigger to we can build a drug to fix it.
It didn't go smoothly.
It resulted in an absolute bloodbath for pharmaceutical companies. The subsequent decades are just a list of catastrophic failures.
The long list.
Let's talk about bapineuzumab. It was one of the early heavy hitters. They run a sweeping phase three trial, published the data around 2014, testing it on both APOE4 gene carriers and non-carriers.
And the result?
Complete clinical failure. No benefit whatsoever.
Precisely. And the failure wasn't just a slight miss. It forced the scientific community to question the very foundation of their understanding.
Which had to be terrifying for the field.
It was a crisis. Bapineuzumab was a monoclonal antibody designed to bind to amyloid and signal the brain's immune system to clear it out. But it failed because of how we fundamentally misunderstood the target.
Right, because then you look at solanezumab, which was tested in the Expedition 3 trial. That read out in 2016.
Solanezumab was designed differently.
Right, it targeted soluble monomeric amyloid. So, like the individual protein strands floating in the brain fluid before they clump together.
Yes, monomers.
And again, it completely missed its primary clinical endpoints. So, my question for you is, why did the scientific community stubbornly keep pouring billions of dollars into this exact same target when the clinical trials were screaming that it wasn't working?
Well, it's easy to look back and incite the sunk cost fallacy, you know? And certainly, there was financial momentum.
Pharma companies had a lot riding on it.
They did. But scientifically, the genetic evidence was just too strong to ignore. The researchers didn't abandon the hypothesis. Instead, they refined their understanding of the mechanics.
They pivoted.
They developed a twofold working theory to explain this graveyard of failed drugs. The first part of that theory was simply an issue of timing.
Meaning they were trying to put out the fire long after the house had burned down.
That is the perfect analogy. Those early trials enrolled patients who already exhibited moderate to severe dementia.
So, the damage was already done.
If we look at the cellular level, by the time someone is in the moderate stages of Alzheimer's, the neurodegeneration is profound. The synapses, the connections between neurons, have physically deteriorated.
So, you can't just reverse that by cleaning up the mess.
Right. Whole networks of brain tissue have atrophied. Pulling amyloid out of an atrophied brain is biologically futile. The structural damage is entirely irreversible at that point.
So, the intervention was simply way too late. But the second part of their theory is what I find fascinating because it gets into the actual architecture of the protein.
Yes, the species of amyloid.
Right. The theory was that these early drugs were attacking the wrong species.
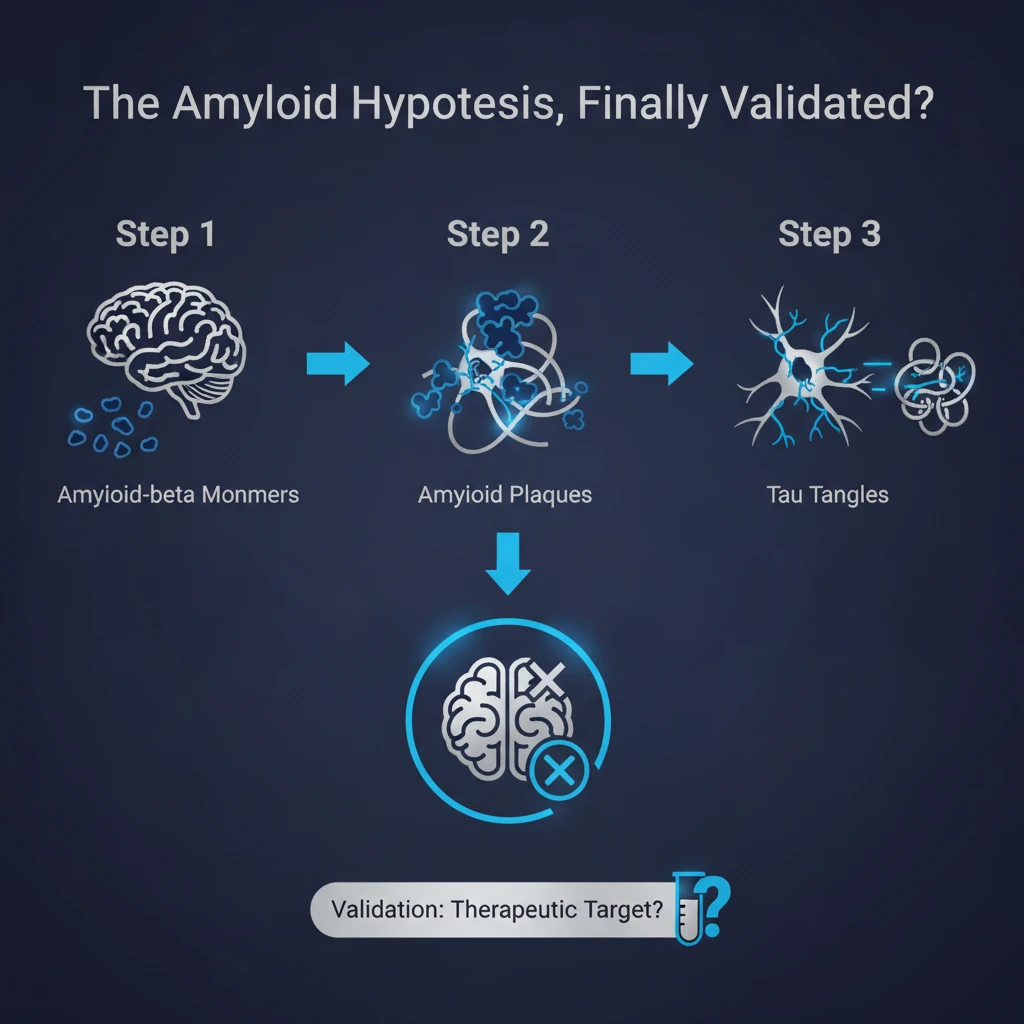
This is a really crucial distinction to understand. Amyloid beta is not a monolithic entity. It undergoes a structural evolution.
Walk us through that evolution.
It starts as monomers. Single, floating protein fragments that get cleaved from the larger precursor protein.
And those monomers are harmless, right?
Generally considered inert. Yeah. They might even have normal physiological functions. But in Alzheimer's, these monomers begin to misfold and aggregate.
They start clumping up.
Exactly. They clump together into oligomers, then longer chains called protofibrils, and finally they deposit into those dense, insoluble plaques.
Okay, let me try an analogy here to see if I have the mechanics right for our listeners.
Let's hear it.
If we think of amyloid buildup like an obstruction on a vital neural highway, solanezumab was targeting the monomers.
Right.
So, that's like a street sweeper trying to capture individual microscopic pieces of gravel scattered in the wind. It's completely ignoring the actual structural blockages. The aggregated oligomers and protofibrils that are physically wedged into the synaptic clefts, disrupting the chemical signaling between neurons.
Then it's highly accurate. The soluble oligomers and protofibrils are the highly neurotoxic species.
They're the real bad guys.
They don't just sit there passively. They actively disrupt synaptic function. They induce oxidative stress and they trigger inflammatory responses from the brain's immune cells, the microglia.
So, the early drugs were essentially missing the target.
Exactly. They were either binding to the harmless monomers, acting like a sponge that got saturated before doing any good, or they were binding inefficiently to the hardened plaques while leaving the toxic oligomers completely free to roam.
Wow, which brings us back to that visceral opening I mentioned. The field is suffering an existential crisis of confidence.
Morale was at rock bottom.
And then a drug comes along that forces regulators into an impossible corner. We really have to dissect the Aducanumab controversy.
Aducanumab, marketed as Aduhelm. This is where the story shifts from a purely biological puzzle to a sweeping regulatory battle.
A very messy battle.
Very. Mechanistically, Aducanumab was designed to fix the targeting error of the earlier drugs. It specifically targeted the aggregated forms of amyloid, the toxic oligomers and the protofibrils.
But the timeline of its clinical trials is what makes this so wild.
It's unprecedented.
Right. So, the manufacturer, Biogen, ran two parallel phase three trials, Emerge and Engage. And in early 2019, they conducted a futility analysis.
Which is a standard procedure.
Yeah, for those tracking the trial designs, that is an independent review conducted midway through a trial by a data monitoring committee.
Right, they look at the unblinded data.
To see if there is any statistical trajectory showing the drug might work. And if the trajectory is flat, they halt the trial.
Which is exactly what happened.
Both Emerge and Engage failed the futility analysis.
The independent committee saw no clinical benefit. So, Biogen halted the program. And the entire neurological community took a collective breath, assuming the amyloid hypothesis had finally sustained its fatal blow.
It was over. But then Biogen did something statistically controversial.
Very controversial.
They went back and performed a post-hoc re-analysis of the data.
Yeah.
They sliced the data differently, looking specifically at a subset of patients who received a higher dose of the drug for a longer period in just one of the trials, the Emerge trial.
Yes.
And in that specific slice of data, they found a positive signal.
From a purely statistical standpoint, post-hoc re-analysis is heavily scrutinized. It is often compared to the Texas Sharpshooter Fallacy.
Firing a shotgun at a barn and then painting a bull's-eye around the tightest cluster of bullet holes.
Exactly. You are finding a pattern after the fact, rather than proving a hypothesis you set out to test.
Right. Despite that, based on that sliced-up data, the FDA made their move. In June 2021, they granted Aducanumab accelerated approval.
Which sent shockwaves through the field.
I bet. But here is a critical detail. They did not approve it because Biogen definitively proved it saved people's memory.
No, they didn't.
They granted the approval based entirely on a surrogate endpoint. We really need to clarify exactly what that means in regulatory science.
A surrogate endpoint is a biomarker. It's a measurable physical sign that is believed to predict a clinical benefit, even if the clinical benefit itself hasn't been definitively proven in the trial.
Can you give an example?
Sure, a classic example in medicine is using viral load as a surrogate endpoint for HIV drugs.
Hmm.
You measure the drop in the virus in the blood, assuming it will prevent the onset of AIDS, rather than waiting years to see if the patient actually develops AIDS.
Got it. So, what was it for this Alzheimer's drug?
In the case of Aducanumab, the surrogate endpoint was the clearance of amyloid plaque from the brain as measured by a PET scan.
And the PET scans were undeniable, right?
The drug acted like a molecular vacuum cleaner. It cleared the plaque beautifully.
But the fallout from using that surrogate endpoint to justify an Alzheimer's drug approval was catastrophic.
It was a massive backlash.
That FDA independent advisory committee we mentioned at the start, they voted 10 to 0 against approval because they felt the clinical data on memory preservation was entirely unconvincing.
And three of those experts resigned in protest after the FDA bypassed their recommendation.
That is just wild.
It ruptured the fundamental trust between regulatory agencies and the clinical community. Yeah. Neurologists rely on the FDA to validate efficacy. When the FDA approved a drug based on biological mechanism rather than a clinical outcome against the advice of its own experts, it placed an impossible burden on individual doctors.
I genuinely struggle to understand the FDA's rationale here. I mean, why didn't they just tell the manufacturer, go run another trial, prove it works on memory, and then we will approve it. Why risk the agency's credibility?
To understand the FDA's action, you really have to look at the systemic pressures operating in the background. Alzheimer's is a terminal, structurally devastating disease with zero disease modifying treatments available at the time.
So, the desperation factor.
Exactly. You have patient advocacy groups and millions of families facing a grim, inevitable decline, and they are essentially demanding access to anything that shows biological activity.
Even if the memory data wasn't there yet.
Right. The accelerated approval pathway was explicitly designed for severe diseases with unmet needs. The regulators looked at the undeniable biological effect, the clearance of the plaque, and made a highly subjective judgment call that it was reasonably likely to predict a clinical benefit down the line.
But the market ultimately rendered its own verdict.
It did indeed. By early 2024, Aducanumab was essentially withdrawn. It was a complete commercial collapse. Doctors wouldn't prescribe it, insurers balked at the cost, and the confirmatory trials were lagging.
However, mathematically and scientifically, Aducanumab was not a failure.
No.
No, because it proved a vital mechanism. It proved that administering a synthetic antibody could successfully cross the blood-brain barrier, locate aggregated amyloid and stimulate the brain's immune system to clear it out.
So, it proved the target was druggable.
Exactly. But it set a relentless new standard. The next drug in line had to prove, unequivocally, that clearing that plaque translated to preserving a patient's memory.
Which brings us to the breakthrough. The moment the 30-year puzzle finally clicked into place.
This is the exciting part.
Out of the ashes of the Aducanumab controversy comes lecanemab, known commercially as Leqembi.
This is the real turning point in modern neurology.
Let's talk about it. Lecanemab's specific biological target is the amyloid protofibril. As we discussed, these are the highly toxic soluble intermediaries.
Yes.
It completely ignores the harmless monomers and hones right in on the most destructive element of the cascade. The pivotal trial that proved its efficacy was called Clarity AD, published in the New England Journal of Medicine in late 2022.
A massively important publication.
It was a rigorously designed trial with nearly 1,800 participants.
And the patient selection methodology here was fundamentally different from the dark ages of those early trials.
They learned from their mistakes.
They entirely avoided the timing mistake. They explicitly enrolled patients with early symptomatic Alzheimer's disease, specifically those diagnosed with mild cognitive impairment or mild Alzheimer's dementia.
So, catching it early.
Yes. And furthermore, every single patient had to have biologically confirmed amyloid pathology via a PET scan or a cerebrospinal fluid analysis before entering the trial.
They weren't guessing anymore.
Exactly. They knew the amyloid was there.
The intervention was intense, though. Patients received intravenous infusions of 10 milligrams per kilogram of the drug every two weeks for 18 months.
It's a significant commitment.
Let's break down exactly what happened because this is where the hypothesis becomes reality. Biologically, the drug was a triumph.
Unquestionably.
By week 79 of the trial, 86% of the patients receiving lecanemab converted to amyloid negative status on their PET scans. Their brains were essentially scrubbed clean of the pathology.
It's an incredible visual on the scans.
But here is the data point that changed history. Clinically, the patients on lecanemab declined 27% less than the patients on the placebo over those 18 months.
And because of that clinical data, the FDA granted it full traditional approval. No surrogate endpoint shortcuts this time. Full approval.
So, let's talk about what declining 27% less actually means.
Right, we have to evaluate what that means for a human being, not just a spreadsheet. We have to interrogate the primary clinical endpoint used in the trial. They used a metric called the CDRSB, which stands for the clinical dementia rating sum of boxes.
Let me stop you right there.
Sure.
Because when I look at the raw math of the CDRSB, I find myself deeply skeptical of the celebration.
A lot of people did.
The CDRSB is an 18-point scale assessing various cognitive and functional domains, you know, memory, orientation, personal care. Zero means perfectly normal, 18 means severe, late stage dementia. The absolute difference between the lecanemab group and the placebo group at the end of 18 months was 0.45 points, less than half a single point on an 18-point scale.
It sounds incredibly small.
In the historical literature of neurology, the minimum clinically important difference, the threshold where a patient actually feels a subjective change, is generally considered to be one to two points. So, how can anyone look at a 0.45 difference and call it a monumental breakthrough? It sounds indistinguishable from a statistical rounding error.
Your skepticism mirrors the exact debate that consumed the neurological community immediately following the data release.
I'm glad I'm not the only one.
If you evaluate a 0.45 point difference in a static vacuum, it absolutely fails to cross that historical threshold of a clinically meaningful change. But Alzheimer's disease is not static. It is a relentless, compounding slope of neurodegeneration.
It accelerates as it goes.
Precisely. If you are slowing that rate of decline by 27% very early in the disease process, that 0.45 absolute difference might translate to preserving a few extra months of independence.
Which is huge for a family.
It might mean a patient can safely manage their own finances, or drive a car, or recognize a spouse for six months longer than they otherwise would have. And, more importantly, we have to look at the mathematical compounding.
Okay, lay that out for us.
If you maintain that 27% reduction in the rate of decline over three, four, five years, the area between the two curves widens dramatically. You are fundamentally altering the trajectory of a terminal disease.
Okay, looking at it through the lens of compounding trajectories over years makes the clinical value much clearer.
It's about buying time.
But there is another statistical metric used in population health that we need to examine, the NNT, the number needed to treat.
Ah, yes. The NNT is a vital concept in health economics. It asks a highly practical question. How many individual patients do you need to treat with this drug to prevent one single patient from crossing a defined threshold of clinical decline over a specific period?
For lecanemab, the estimates are quite high, right?
Yes, depending on the specific cognitive scale you are looking at, you have to treat between 10 to over 20 patients to see that meaningful, noticeable benefit in just one of them over an 18-month period.
While that sounds high to a layperson, it is actually quite standard for preventative interventions in chronic diseases, isn't it?
It is. Consider the NNT for prescribing statins to prevent a primary cardiovascular event. You are treating dozens of people to prevent one heart attack.
That puts it in perspective.
However, it becomes an immense challenge in the clinic. A neurologist has to sit across from a patient and explain that while the biological clearance of plaque is almost guaranteed, the noticeable clinical benefit for them as an individual is a statistical probability, not a certainty.
Man, that's a tough conversation.
Very tough.
And just as the medical community is digesting the continuous two-week infusion protocol of lecanemab, a second drug arrives that completely upends the treatment model.
It really shook things up.
It introduces the concept of finite dosing. We are talking about donanemab, marketed as Taisumra.
Donanemab represents a significant structural evolution within this new class of anti-amyloid therapies. It targets a highly specific post-translationally modified form of amyloid beta, known as N3PE A-beta.
Let's translate the biology there. Post-translationally modified means the protein is altered after it is initially created by the cell.
Right.
But the crucial detail isn't just the chemical structure, it is where this specific protein lives. This modified amyloid is found exclusively in the hardened core of the established amyloid plaques. It is not circulating in the soluble fluid.
It's essentially a targeted mechanism designed to break down the hardest, most entrenched structural blockages. It binds to the core and recruits the microglia to dismantle the plaque entirely.
The clinical trial for donanemab was called TRAILBLAZER-ALZ 2, published in JAMA in mid-2023, enrolling over 1,700 participants. It received full FDA approval in July 2024.
Yes, the momentum was building.
The administration is an IV infusion every four weeks. But the protocol is what is truly revolutionary here. With lecanemab, it's a chronic maintenance drug. You take it perpetually.
Right.
But with donanemab, the protocol requires serial PET scans. Once the imaging confirms that the amyloid burden in the brain has dropped below a specific visual threshold, meaning the plaques are functionally gone, the treatment is halted.
And the efficiency was remarkable. The median time it took for patients in the trial to reach that threshold of amyloid clearance was roughly 12 months.
Just one year.
After that point, they were transitioned off the active drug and onto a placebo. And the critical finding was that the clinical benefit, the slowing of the cognitive decline, was maintained even after the antibody was no longer being pumped into their system.
The analogy I keep coming back to is the difference between a daily maintenance medication and calling an exterminator.
I like this analogy.
Lecanemab is like taking medication every single day to manage a chronic condition. Donanemab acts like an exterminator. You blast the environment until the infestation is completely eradicated, and then the exterminator packs up their equipment and leaves.
Your house is safe.
The structural integrity of the house remains protected precisely because the destructive element has been removed.
That is an excellent way to conceptualize the clinical utility. The ability to halt treatment has sweeping implications.
Financially and physically.
Exactly. Not only does it vastly reduce the logistical burden on infusion centers, but it mitigates the cumulative risk of severe side effects associated with long-term antibody exposure.
Which we will get into.
But, the TRAILBLAZER-ALZ 2 trial contributed something even more profound to our understanding of the biology because they stratified their clinical results based on the presence of a second pathological protein.
Ah, tau.
Yes, tau.
This is where the mechanisms of the disease really come into focus. They looked at patients who had low to medium levels of tau accumulation, indicating an earlier phase of the disease process.
Right.
In that specific subgroup, donanemab slowed cognitive decline by 35% on a metric called the iADRS and 36% on our familiar CDRSB scale. That translates to an absolute difference of about 0.7 points on the CDRSB, which is a stronger signal than we saw with lecanemab.
A very strong signal.
But then they looked at the patients who had high tau pathology. What happened there?
The clinical benefit in the high tau group was numerically smaller and less statistically robust. To understand why, we really have to look at the cellular architecture.
Break it down for us.
Tau is a protein that stabilizes the microtubules inside the neurons. It acts as the internal skeleton of the cell. In Alzheimer's, after the amyloid plaques begin to accumulate outside the cells, a secondary cascade is triggered.
A chain reaction.
Exactly. The tau proteins inside the cells become hyperphosphorylated. They detach from the microtubules, they clump together into neurofibrillary tangles, and the internal skeleton of the neuron collapses. The cell dies.
So, the amyloid is the match, but the tau is the fire.
Exactly. And once those tau tangles begin to propagate, they spread from neuron to neuron like an infection.
Wow.
The current biological consensus is that if a patient has high levels of tau, the neurodegenerative fire is already burning out of control.
So, removing the match at that point.
Doesn't put out the fire. At that point, going back and pulling the initial amyloid trigger out of the brain has rapidly diminishing clinical returns. The destruction is self-perpetuating.
That dynamic definitively proves that these anti-amyloid therapies are intensely, uncompromisingly time-sensitive.
The window is so narrow.
If you wait until the tau pathology is widespread, the structural damage is done. And that brings us to the most urgent logistical hurdle in modern medicine.
The bottleneck.
Right, we have two validated therapies capable of altering the course of Alzheimer's disease. But knowing how to put out the fire only matters if you can find the burning houses before the structural collapse. How do neurologists identify these early stage patients without collapsing the health care infrastructure?
This is the diagnostic bottleneck. The inclusion criteria for these therapies are exceptionally rigid. You must find patients in the early symptomatic phase. Mild cognitive impairment.
Which can be hard to distinguish from normal aging?
Yes. Patients who have progressed to moderate or severe dementia are explicitly excluded from the drug labels because there is no biological evidence that they will benefit, and they would still be exposed to all the severe risks.
But historically, distinguishing between normal age-related cognitive slowing and early stage Alzheimer's required highly invasive or intensely expensive procedures.
You can't just guess.
No, you can't visually diagnose an amyloid plaque. You either needed a PET scan, which requires specialized radioactive tracers manufactured in a cyclotron and costs thousands of dollars.
If you can even find a facility that does it.
Right. Or you needed to perform a lumbar puncture, a spinal tap to extract cerebrospinal fluid and measure the amyloid levels directly.
Both of those diagnostic tools are highly sensitive and specific, but they are fundamentally unscalable for a disease that impacts tens of millions of people globally.
It's just not practical.
You cannot funnel the entire aging population of a country through specialized PET scanners or subject them to spinal taps simply to screen them for drug eligibility. The infrastructure would shatter.
But the clinical landscape is currently undergoing a diagnostic revolution parallel to the drug approvals, right? We are transitioning away from the scanner and the spinal needle and moving toward blood tests.
The rapid maturation of blood-based biomarkers is perhaps the most critical unsung hero of this entire era. We are specifically looking at a plasma biomarker called P-tau 217.
Let's break down how this works mechanically. Advanced assay platforms, utilizing technologies like Quanterix's Simoa, which is capable of single molecule array detection, can now isolate microscopic traces of this specific phosphorylated tau protein that is leaked across the blood-brain barrier and into the peripheral bloodstream.
It's incredible technology.
And these platforms are demonstrating sensitivity and specificity rates exceeding 90%. That means a peripheral blood draw in a standard clinic can match the diagnostic accuracy of a specialized brain scan in identifying amyloid pathology.
It acts as an unprecedented mechanism for clearing the bottleneck. The future clinical pathway is highly streamlined.
How does that look for a normal patient?
Well, a primary care physician evaluates a patient presenting with mild cognitive decline. Instead of immediately referring them to a six-month wait list for a specialized neurologist and a PET scan, they order a routine blood draw for plasma P-tau 217.
Simple.
If the biomarker is elevated, then the patient is triaged into the specialized system for a confirmatory PET scan or lumbar puncture before initiating the intravenous therapy. It democratizes the initial screening process.
But once a patient clears that initial diagnostic hurdle, they immediately slam into a highly complex genetic filter.
Yes, the genetics.
The FDA prescribing labels for both lecanemab and donanemab strongly recommend or mandate genetic testing before initiating therapy. They are specifically looking at the APOE gene. Why is the patient's genotype the ultimate gatekeeper for these drugs?
The genetic testing is entirely driven by the profound safety risks associated with the therapy. We're talking about ARIA. Amyloid-related imaging abnormalities.
Right.
ARIA is the defining severe adverse event of the anti-amyloid drug class. It manifests in two primary forms. ARIA-E, which is vasogenic edema or swelling in the brain tissue. And ARIA-H, which involves microhemorrhages or localized bleeding in the brain.
Why does pulling plaque out of the brain cause bleeding? Like, what is the physical mechanism there?
Because amyloid doesn't just deposit in the tissue between the neurons. In many Alzheimer's patients, amyloid also deposits heavily into the walls of the blood vessels in the brain. A condition called cerebral amyloid angiopathy.
Oh, wow.
The amyloid protein effectively replaces the smooth muscle of the vessel wall. When you pump high doses of an antibody into the bloodstream, it attacks the amyloid in those blood vessels. As the antibody breaks down the amyloid, it fundamentally compromises the structural integrity of the vessel wall.
The wall gets weak.
The vessel becomes leaky, resulting in fluid seeping into the brain tissue causing swelling, or red blood cells leaking out, causing microhemorrhages.
That is terrifying. It is literally like pulling load-bearing pillars out of a crumbling building, and the APOE4 gene accelerates this risk.
Dramatically. The APOE gene regulates lipid transport in the brain. But the E4 variant is highly correlated with aggressive amyloid accumulation, particularly in the blood vessels.
Okay, so if you have that gene.
Patients who are homozygous APOE4 carriers, meaning they inherited the E4 allele from both their mother and their father, face an approximately four-fold higher risk of experiencing severe ARIA compared to non-carriers.
Four times the risk of brain bleeding.
The risk profile is so extreme that many clinical practice guidelines suggest neurologists should explicitly avoid treating homozygous carriers, or do so only under the most intense frequent MRI surveillance.
Furthermore, the clinical contraindications go far beyond genetics. If a patient has a history of prior microhemorrhages, a recent stroke, or if they are currently taking anticoagulant medications, blood thinners, they are almost universally excluded from treatment due to the compounding risk of catastrophic brain bleeding.
That rules out a lot of people.
So, to contextualize this for you listening, if you are experiencing mild cognitive impairment, you cannot simply demand a blood test and expect a prescription.
Absolutely not. The intersection of the strict biological window, the genetic risk factors, and the systemic contraindications creates an intensely difficult clinical reality.
It's a massive filter.
The blood-based biomarkers make it incredibly efficient to identify patients who have the pathology. But the safety profile of the drugs means clinicians are forced to exclude a substantial proportion of those exact patients.
Which transitions us to the final and undoubtedly the most sobering segment of our analysis. We have to leave the sterile, tightly controlled environment of statistical trial data and confront the messy, underfunded reality of the global health care ecosystem. We are looking at the bedside reality.
Integrating a fundamentally new, highly complex treatment modality into existing health care systems is a logistical undertaking of the highest order.
The global landscape for access is currently highly fractured. In the United States, Medicare has agreed to cover both lecanemab and donanemab, though they enforce stringent prior authorization requirements and mandate physician enrollment in data registries.
Yes, the U.S. is moving forward, albeit cautiously.
However, you look across the Atlantic to the United Kingdom, and the reality is entirely different. In early 2024, the UK's National Institute for Health and Care Excellence, commonly known as NICE, issued draft guidance, explicitly recommending against the use of lecanemab in the National Health Service.
Major decision.
They concluded that the drug does not represent a cost-effective use of public health care resources. Meanwhile, the European Medicines Agency grants authorization, but individual member states dictate actual access.
It is vital to analyze the underlying logic of the NICE decision. Their rejection was not a repudiation of the clinical trial data.
They weren't saying it doesn't work.
Exactly. They acknowledge the drug demonstrates biological efficacy and slows clinical decline. Their decision was driven entirely by health economics and infrastructure capacity. They use a metric called quality-adjusted life years or QALYs to measure cost-effectiveness.
How does that math work when applying it to this specific treatment protocol?
You have to aggregate the total systemic cost of delivering the therapy. You are not just paying tens of thousands of dollars annually for the drug itself. You are paying for the initial PET scans or lumbar punctures. You are paying for the genetic sequencing.
The blood tests, the lab work.
The patient requires specialized infusion chairs every two to four weeks. Crucially, because the risk of ARIA, the protocol mandates serial MRI scans, often three or four scans within the first year of treatment, simply to monitor for asymptomatic brain bleeding.
That's a massive resource drain.
NICE modeled the economic weight of hundreds of thousands of eligible patients requiring serial MRIs, specialized neurological consultations, and constant infusion center access. And they concluded that the fractional slowing of cognitive decline did not mathematically justify paralyzing the broader health care infrastructure.
When you lay out the logistical reality like that, it illuminates the immense emotional burden placed on the individual clinician. Think about the dynamics in a neurologist's office right now.
So wrenching.
A family arrives, desperate and terrified. They have read an optimistic article declaring that a cure for Alzheimer's has finally been approved by the FDA. The doctor has to absorb that hope and methodically dismantle it.
Yes.
They have to explain, we cannot stop the disease. We can theoretically slow the rate of your cognitive decline by roughly 27% to 35%. To achieve that, you must undergo regular IV infusions. We have to monitor your brain for potentially fatal swelling and bleeding, and ultimately, due to your genetic profile or your cardiovascular medication, I cannot prescribe it to you at all.
It completely redefines the role of the modern neurologist. The core clinical skill in 2026 is no longer focused solely on achieving an accurate diagnosis.
Managing expectations.
It is the art of shared decision-making. Clinicians must navigate the profound disconnect between the publicized scientific breakthroughs and the nuanced, often restrictive, clinical realities. They have to communicate to genuine, validated efficacy of these therapies without allowing patients to succumb to false hope regarding the ultimate trajectory of the disease.
The macro meets the micro.
The economic and infrastructure modeling we see from agencies like NICE is simply the macro level reflection of those brutal micro level conversations happening in clinics every single day.
So, how do we synthesize this monumental shift in neuroscience? As we conclude this deep dive, let's look at the totality of the landscape.
It's a lot to take in.
The amyloid hypothesis has successfully navigated the transition from a heavily debated, heavily funded theoretical framework into a clinically validated, actionable reality. The 30-year biological puzzle has yielded tangible results.
Which is worth celebrating.
We have two distinct therapies, lecanemab and donanemab, that prove altering the amyloid cascade fundamentally slows the progression of Alzheimer's disease. Furthermore, the advent of highly accurate blood-based biomarkers like P-tau 217 is revolutionizing the diagnostic pathway.
Effectively eliminating the historic reliance on unscalable PET scans and lumbar punctures for initial screening.
Right. The scientific community has established the definitive foundation upon which all future disease modifying therapies will be built. The clinical benefit, while modest, is undeniably real and represents a structural change in how we combat neurodegeneration.
And to you, the listener, the evolution of this field demands your attention, regardless of your immediate proximity to the disease. As the demographic reality of an aging global population accelerates, the mechanisms our societies use to absorb this infrastructure shock, how we determine genetic eligibility, how we finance the serial imaging and infusion logistics, and how we allocate specialized clinical resources will generate economic ripples that impact every single facet of global health care.
It is the ultimate test of how modern medicine scales complex biological interventions across a population.
I want to leave you with one final, deeply provocative biological question that emerges from the exact clinical data we just dissected.
I know where you're going with this.
If administering a targeted monoclonal antibody effectively clears nearly 100% of the toxic amyloid plaques from the brain, yet that profound biological clearance only translates to a 27% to 35% slowing of the clinical cognitive decline. What biological mechanism is driving the remaining 65% of the neurodegeneration?
That is the million dollar question.
We have definitively proven that amyloid is the pathological trigger. We pulled the trigger out and the progression slowed. But if the amyloid is only the match, what constitutes the rest of the inferno?
Is it the tau?
Right. Are the intracellular tau tangles operating autonomously once initiated? Is the brain's immune system, the microglia, trapped in a self-perpetuating cycle of neuroinflammation and synaptic pruning?
There are so many variables.
The data clearly dictates that the next frontiers of neuroscience lie in uncovering those secondary cascades. The scientific community has finally, agonizingly, locked the first critical piece of the puzzle into place. But the true nature of the remaining picture, that is the mystery still waiting to be solved.
Free Clinical Resources
Episode Infographic
Key data, mechanisms, and clinical takeaways distilled into a single shareable page. Save it for reference or use it in your next team discussion.
Clinical Flashcards
Core concepts from this episode in flashcard format, structured for spaced repetition, pre-consultation review, or CPD reflection with your team.
More from: Dementia Frontiers Deep Dive Series
Why New Alzheimer's Drugs Make Brains Bleed?
ARIA (amyloid-related imaging abnormalities) affects up to 24% of patients on anti-amyloid therapy. APOE4 genotyping, MRI monitoring protocols, severity-graded management, and the anticoagulation dilemma: a practical guide to navigating the risk.
Targeting Tau Inflammation And Alzheimer's Metabolism
Amyloid clearance is only one part of the Alzheimer's story. Why tau remains the closer correlate of cognitive decline, what the TREM2 biology tells us about microglia, what the GLP-1 signal means, and how platform trials are moving toward multi-target combination therapy.

Preventing 45 Percent Of Dementia Cases
The Lancet Commission 2024 estimates 45% of dementia is preventable. SPRINT-MIND showed 19% MCI reduction from intensive blood pressure control. FINGER showed 25% better cognitive scores with multi-domain lifestyle intervention. ACHIEVE showed 48% slowing in high-risk patients with hearing aids. The evidence is here and we walk through it.
ART-2026-351
·07/26
Drafted with AI assistance, reviewed and approved by the editorial team. This publication is intended for healthcare professionals, researchers, and life science industry professionals. Content is provided for informational and educational purposes only and does not constitute medical advice.

Digital health and patient experience are my beat: the apps, the wearables, the real-world evidence claims, and whether any of it changes outcomes. Sceptical by training and optimistic by instinct.
Cite This Podcast
Ward S, Reeves T. The amyloid hypothesis, finally validated?. The Life Science Feed. Published June 11, 2026. Updated July 26, 2026. Accessed July 27, 2026. https://thelifesciencefeed.com/neurology/alzheimer-disease/research/amyloid-hypothesis-finally-validated-dementia-ep1.
Editorial & AI Standards
All content is researched from peer-reviewed, open-access sources: published trial data, clinical guidelines, and regulatory filings. AI tools are used solely to structure and summarise that evidence; no AI-generated conclusions appear without editor verification against the primary source.
Every article is reviewed by a named editor before publication. Source citations are listed in the References section. This content does not represent the views of any pharmaceutical company, medical device manufacturer, or healthcare provider.
Licence & Rights
© 2026 The Life Science Feed. All rights reserved. Unless otherwise indicated, all content is the property of The Life Science Feed and may not be reproduced, distributed, or transmitted in any form or by any means without prior written permission.
Podcast Disclaimer
This podcast is produced for educational and informational purposes only. The conversation between hosts represents a discussion of published clinical evidence and is not intended as clinical advice, a substitute for professional medical judgment, or a recommendation for any specific treatment. Healthcare professionals should rely on their own clinical training, current guidelines, and individual patient assessment when making treatment decisions. The views expressed are those of the hosts and do not constitute endorsement of any specific therapy, product, or manufacturer.
References
Budd Haeberlein S et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer's Disease. J Prev Alzheimers Dis. 2022;9(2):197–210.
Doody RS et al. Phase 3 Trials of Solanezumab for Mild-to-Moderate Alzheimer's Disease. N Engl J Med. 2014;370:311–321.
Hardy J, Higgins G. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi:10.1126/science.1566067
National Institute for Health and Care Excellence. Lecanemab for treating mild cognitive impairment or mild dementia caused by Alzheimer's disease: NICE technology appraisal guidance [draft]. 2024.
Palmqvist S et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA. 2020;324(8):772–781. doi:10.1001/jama.2020.12134
Salloway S et al. Two Phase 3 Trials of Bapineuzumab in Mild-to-Moderate Alzheimer's Disease. N Engl J Med. 2014;370:322–333.
Sims JR et al. Donanemab in Early Symptomatic Alzheimer's Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA. 2023;330(6):512–527.
van Dyck CH et al. Lecanemab in Early Alzheimer's Disease. N Engl J Med. 2023;388:9–21.